
译自:河南农业大学兽医学院Jinyuan Li
摘要
猪流行性腹泻病毒(PEDV)是引起猪流行性腹泻(PED)的病原体,会导致猪只呕吐、水样腹泻和严重脱水。该病毒对新生仔猪表现出极高的致死率,对全球养猪业构成重大威胁并造成巨大的经济损失。宿主先天免疫系统是抵御病毒入侵的主要防线;然而,PEDV采用多种策略来逃逸这种免疫反应。本综述系统总结了PEDV逃逸宿主先天免疫的多种分子机制,包括通过病毒编码的蛋白干扰宿主细胞内信号通路、拮抗宿主的抗病毒因子及相关免疫基因以抑制先天免疫反应、以及调控宿主细胞的自噬过程,从而实现逃逸宿主先天免疫。全面理解PEDV如何破坏先天免疫对于制定有效的控制策略和治疗方法至关重要。本综述旨在为抗击PED提供新的见解和潜在靶点。
关键词:
PEDV;PED;先天免疫;免疫逃逸;控制策略与治疗方法
1.引言
猪流行性腹泻(PED)是一种主要由猪流行性腹泻病毒(PEDV)引起的急性肠道疾病,其特征是腹泻、呕吐、采食量减少和脱水。虽然所有年龄和品种的猪均易感,但该病毒对14日龄内的仔猪威胁最为严重,极大地危害养猪业并在全球范围内造成巨大的经济损失。PEDV属于甲型冠状病毒属,是一种有包膜的单股正链RNA病毒。其基因组大小约为28 kb,包含一个5'非翻译区(UTR)、至少七个开放阅读框(ORF1a,ORF1b,以及ORF2至ORF6)和一个3'UTR。ORF1a和ORF1b代表主要的非结构蛋白(NSP)编码区。这两个ORF之间存在46个核苷酸的重叠,促进了核糖体移码,从而能够翻译出一个大的多聚蛋白(pp1ab)。随后,pp1ab通过蛋白水解切割产生16个NSP(NSP1至NSP16)。靠近3'末端的其余ORF(ORF2至ORF6)编码四种结构蛋白:刺突蛋白(S)、包膜蛋白(E)、膜蛋白(M)和核衣壳蛋白(N)。
宿主进化出了一个复杂的免疫系统,包括先天性和适应性免疫两部分,以应对病原体威胁。先天免疫系统通过物理化学屏障——包括皮肤、黏膜表面以及酶和补体蛋白等化学物质——作为第一道防线,阻止微生物入侵。病原体突破屏障后,受感染细胞释放细胞因子和趋化因子启动炎症反应,招募吞噬细胞和抗原呈递细胞(APC)以清除病原体。对于逃逸这些机制的病原体,适应性免疫系统通过APC介导的MHC分子抗原呈递被激活,触发病原体特异性的T和B淋巴细胞反应,产生中和抗体并建立免疫记忆。这两个层级提供了广谱、快速的保护,在感染早期主要由先天免疫介导。作为专性细胞内病原体,病毒被宿主模式识别受体(PRR)检测到,这些受体识别病原体相关分子模式(PAMP),例如病毒核酸,从而诱导干扰素(IFN)和促炎细胞因子的产生,以建立抗病毒状态。然而,包括PEDV在内的病毒已经进化出对抗策略来破坏先天免疫信号传导,从而促进病毒复制和致病机制。
本综述系统阐述了PEDV逃逸宿主先天免疫的多种分子机制。PEDV通过其结构蛋白、非结构蛋白和辅助蛋白的协同作用,采用复杂的免疫逃逸策略,通过破坏细胞内信号通路、拮抗抗病毒效应因子和免疫相关基因以及劫持细胞自噬过程,共同削弱宿主先天免疫。值得注意的是,PEDV逃逸宿主先天免疫的能力是其在宿主体内持续存在并在猪群中高效传播的主要原因。因此,阐明这种免疫逃逸的核心机制对于克服当前PED控制策略的瓶颈至关重要。这些机制不同但功能趋同的干预措施使PEDV能够系统性地绕过先天免疫监视,并建立一个有利于病毒复制的细胞环境。阐明这些分子策略不仅增进了我们对冠状病毒致病机制的理解,也为开发有针对性的对策提供了合理的框架,包括蛋白特异性抑制剂、自噬调节化合物以及基于结构的、具有增强效力的疫苗设计,以对抗这种造成巨大经济损失的猪病原体。
2.病毒编码蛋白在PEDV逃逸宿主先天免疫中的作用
2.1.结构蛋白在PEDV逃逸宿主先天免疫中的作用_
2.1.1.S蛋白
PEDV S糖蛋白,作为病毒包膜上最大的结构蛋白,通过一个复杂的两步机制协调病毒进入。这种三聚体糖蛋白由S1(残基1–789)和S2(790–1383)亚基组成,其中S1结构域含有关键的中和表位(包括S1[0],S1[A],COE,SS2,SS6,和C末端表位)以及两个功能不同的受体结合结构域(S1-NTD和S1-CTD),它们共同决定了病毒的抗原性和宿主细胞附着。S1与受体结合后,构象变化暴露出蛋白水解切割位点,导致S1/S2亚基解离,随后由S2介导膜融合——这是一个由融合肽插入宿主膜启动的过程。S蛋白上免疫优势表位的战略性分布,特别是在S1结构域(氨基酸1–219,435–485,499–638,748–755,764–771)和C末端(氨基酸1368–1374),使这种结构蛋白成为基于表位的疫苗设计的首要靶点。这些分子特征不仅阐明了PEDV的入侵策略,也为开发针对这种具有重要经济意义的猪病原体的靶向干预措施提供了结构框架。I型干扰素(IFN-I)是触发宿主抗病毒防御的关键先天免疫细胞因子。从机制上讲,PEDV的S蛋白直接与宿主表皮生长因子受体(EGFR)结合。随后的EGFR激活启动下游JAK2-STAT3信号传导,其中STAT3作为IFN-I产生的转录抑制因子(图1)。这一级联反应因此抑制了IFN-I介导的抗病毒反应。值得注意的是,S1亚基在PEDV感染期间成为细胞凋亡的有效诱导剂。通过促进程序性细胞死亡,S1促进了病毒的释放,并使病毒能够逃逸细胞内免疫监视——这是一种增强病毒传播同时对抗宿主防御机制的策略。同时,S蛋白与EGFR相互作用,抑制黏膜抗病毒细胞因子的产生——特别是关键的黏膜干扰素IFN-λ——从而进一步损害黏膜免疫监视功能。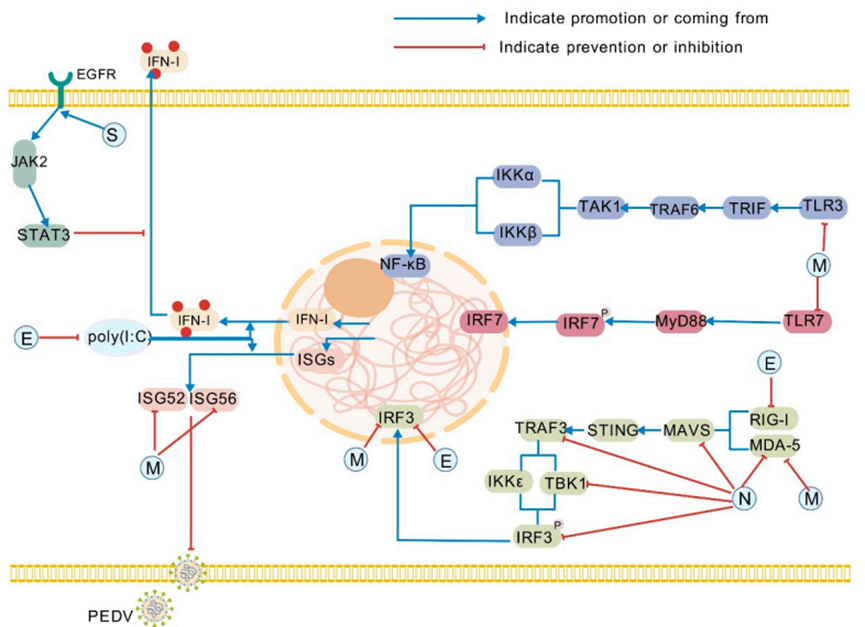
图1:结构蛋白在PEDV逃逸宿主先天免疫中的作用。具体而言,这些结构蛋白抑制JAK2/STAT3、NF-κB和IRF等关键节点,干扰TLR3、TLR7和RIG-I/MDA-5等PRR,并拮抗IFN刺激因子。它们不仅能对宿主先天免疫反应产生拮抗作用,还能通过这种拮抗作用协助病毒逃逸宿主先天免疫防御。EGFR:表皮生长因子受体;poly(I:C):聚肌苷酸-聚胞苷酸;JAK2:Janus激酶2;STAT3:信号转导与转录激活因子3;TLR3:Toll样受体3;MAVS:线粒体抗病毒信号蛋白;IKKα:IκB激酶α;IKKε:核因子κB抑制因子ε激酶;STING:干扰素基因刺激物。该图由BioGDP.com创建。
2.1.2 M蛋白
PEDV M蛋白是一种高度保守的227个氨基酸的III型糖蛋白,在病毒粒子组装和出芽中扮演多重角色,同时也是一个关键的免疫原性结构成分。在感染过程中,M蛋白遍布细胞质,并通过下调细胞周期蛋白A诱导猪肠上皮细胞(IEC)的S期细胞周期阻滞。从机制上讲,M蛋白与真核翻译起始因子3L(eIF3L)相互作用,抑制这种病毒复制的负调控因子以增强PEDV的增殖。此外,M蛋白对包括RIG-I、TLR3、TLR7和干扰素调节因子3(IRF3)在内的先天免疫传感器表现出广泛的拮抗作用。它通过结合IRF7相互作用结构域(ID),特异性靶向IRF7——IFN-I产生的主要转录调节因子。这种相互作用阻碍了TBK1/IKKε介导的磷酸化,从而抑制了IRF7二聚化、核转位以及随后的IFN-I诱导。此外,M蛋白抑制仙台病毒诱导的IFN-β、ISG52和ISG56的表达(图1)。作为这些免疫逃逸策略的补充,M蛋白与热休克蛋白70(HSP70)形成复合物,调节宿主先天免疫和病毒复制效率。
2.1.3 E蛋白
E蛋白是PEDV最小的结构成分(76个氨基酸),促进病毒粒子的组装和出芽,同时表现出战略性的免疫颠覆。E蛋白主要定位于内质网(ER),通过上调葡萄糖调节蛋白78(GRP78)诱导ER应激(ERS)。同时,它激活NF-κB信号并增强白细胞介素-8(IL-8)和抗凋亡蛋白Bcl-2的表达——尽管这些事件可能在感染后期显现。相比之下,感染早期阶段的特点是E介导的NF-κB激活抑制,可能维持病毒复制。作为一个重要的IFN-I拮抗剂,PEDV E蛋白抑制IFN-I的产生。现有证据表明,PEDV的E蛋白可以通过阻断RIG-I介导的信号通路和干扰IRF3的激活来抑制poly(I:C)诱导的IFN-β和ISG的产生(图1)。E蛋白还可以直接结合IRF3,将其滞留在细胞质中并阻断其入核,从而抑制IFN-β的转录。
真核翻译起始在其限速步骤受到四种eIF2α激酶——PKR、PERK、HRI和GCN2——的关键调控,它们通过磷酸化eIF2α来控制蛋白质合成。该过程的核心是eIF4F起始复合物,包括eIF4E、eIF4G和eIF4A,其中eIF4B和多聚腺苷酸结合蛋白(PABP)作为必需的辅助因子。在PEDV感染期间,E蛋白增强GRP78和钙联接蛋白(CANX)的表达,同时增强PKR和PERK的磷酸化。这种双重作用激活了ERS反应的PERK/eIF2α通路,从而抑制宿主蛋白质翻译。从机制上讲,E诱导的eIF2α磷酸化触发了应激颗粒(SG)的形成——这是一种翻译沉默的核糖核蛋白聚集体。ER定位的E蛋白进一步将ER结构重组为点状结构,并上调G3BP1以促进SG组装。至关重要的是,E蛋白的过表达导致全局翻译停滞和内源性蛋白质合成衰减,而不改变mRNA转录水平,证实了翻译(而非转录)抑制。总的来说,PERK/eIF2α激活对于SG生物发生和翻译停滞都是必需的。
2.1.4 N蛋白
PEDV N蛋白是PEDV编码的表达最丰富且高度保守的结构蛋白,主要定位于宿主细胞质,负责协调病毒RNA的转录/复制并调节宿主细胞代谢。此外,N蛋白在宿主-病原体冲突期间采用多方面的策略来颠覆细胞生理学。关键的是,它延长S期进程并抑制宿主细胞增殖——从而为强大的病毒复制创造了有利条件。
同时,N蛋白破坏ER稳态,引发错误折叠蛋白的积累,从而损害ER功能并诱导ERS。它还上调IL-8表达并劫持自噬以促进病毒复制。至关重要的是,N蛋白通过拮抗I型(IFN-I)和III型干扰素(IFN-λ)来执行协调的免疫逃逸。从机制上讲,它直接结合TANK结合激酶1(TBK1),抑制IRF3磷酸化和核转位以抑制IFN-I产生。N蛋白通过对抗IRF3和NF-κB激活进一步阻断IFN-β和ISG表达,同时阻碍RIG-I/MDA5/MAVS/TRAF3介导的IFN-β启动子活性(图1)。矛盾的是,尽管N蛋白通过猪肠道TLR2/3/9通路激活NF-κB,但它通过阻断NF-κB核转位来拮抗IFN-λ的产生。在表观遗传学上,N介导的组蛋白去乙酰化酶1(HDAC1)抑制增强了STAT1乙酰化。这种翻译后修饰抑制了STAT1磷酸化,随后损害了其核转位和抗病毒基因表达——最终实现免疫逃逸。
PEDV的结构蛋白通过协同作用,共同建立了针对宿主先天免疫的多层次防御策略。例如,S蛋白通过利用EGFR-JAK2-STAT3信号轴破坏IFN-I产生,并通过诱导细胞凋亡促进病毒释放。M蛋白靶向干扰素调节因子7(IRF7)和多个PRR,从而破坏IFN介导的信号级联。同时,E蛋白通过激活PERK/eIF2α通路抑制RIG-I信号传导和宿主蛋白质翻译。N蛋白通过阻碍TBK1-IRF3和NF-κB通路来拮抗I型和III型干扰素(IFN-I/λ)。总的来说,这些结构蛋白靶向先天免疫信号传导的关键节点,同时也损害细胞稳态(例如,ER功能、细胞周期进程),从而为病毒复制创造一个优化的环境。
2.2.NSPs在PEDV逃逸宿主先天免疫中的作用
2.2.1 NSP1
从pp1a的N末端加工而来的110个氨基酸的NSP1,定位于宿主线粒体、ER和高尔基体,以促进病毒基因表达。在所有21种PEDV蛋白中,NSP1构成了最有效的IFN拮抗剂。冠状病毒NSP1蛋白——在甲型和乙型冠状病毒中作为属特异性标记物保守存在——尽管序列同一性低,但作为关键的IFN抑制因子发挥作用。至关重要的是,它们共享一个保守的核心结构域,赋予其相似的生物学功能。从机制上讲,SARS-CoV(乙型冠状病毒)NSP1结合40S核糖体亚基,使翻译失活并触发宿主mRNA的内切核酸酶切割。它同时通过抑制IFN产生和先天免疫来抑制抗病毒信号传导。MERS-CoV NSP1表现出类似的宿主基因抑制功能。类似地,PEDV NSP1拮抗先天免疫,其N93/95A突变显著减弱了病毒的适应性:突变体感染的细胞表现出6.8倍升高的IFN-βmRNA、增强的IFN敏感性以及受损的复制效率——证明了NSP1在免疫逃逸中不可或缺的作用。
体内新生仔猪攻毒研究表明,N93/95A突变体能够提供针对致死性感染的完全保护,防止严重的水样腹泻和死亡。这些发现确立了N93/N95残基作为PEDV毒力和免疫逃逸的结构决定因素,其中突变从根本上改变了病毒的致病机制。先前的证据表明,NSP1中包含N93和N95残基的区域可能直接参与结合宿主相关蛋白。N93/95A突变诱导该区域结构改变,损害了其与CREB结合蛋白(CBP)的相互作用。因此,该突变无法有效阻断CBP-IRF3复合物的形成,导致IFN-I和干扰素刺激基因(ISG)的表达上调。这些发现表明,N93和N95残基通过维持NSP1与宿主蛋白之间的正确结合,在NSP1介导的免疫逃逸中发挥关键作用,从而抑制宿主免疫反应。虽然MAVS主要驱动IFN-I诱导,而过氧化物酶体通过过氧化物酶体MAVS依赖性信号传导——由IRF1协调——促进IFN-λ产生,但NSP1通过阻断IRF1核转位和减少过氧化物酶体丰度来抑制IFN-III。除了IFN拮抗作用外,NSP1阻碍NF-κB核转位并抑制IFN-β和促炎细胞因子(TNF-α,IL-1β,IL-6,IL-15,IL-17)的表达。在PEDV感染的LLC-PK1细胞早期,NSP1靶向IκBα磷酸化和泛素化,阻止p65核转位,从而终止NF-κB信号传导——有效地沉默抗病毒细胞因子级联反应(图2)。
最近的证据表明,PEDV NSP1通过靶向NLRC5——MHC-I基因的主要反式激活因子——选择性抑制病毒诱导的MHC-I上调。值得注意的是,NSP1在生理条件下不改变NLRC5或MHC-I的基础表达,表明其专门在病毒感染期间抑制新生NLRC5 mRNA的翻译(图2)。这种靶向抑制使受感染细胞通过减少MHC-I表面表达来模拟未感染的表型,从而逃避免疫监视。由CD8+T细胞和自然杀伤(NK)细胞的监视。因此,受感染细胞逃脱了细胞毒性清除,通过削弱适应性免疫促进了持续性病毒感染。
图2:NSPs和辅助蛋白在PEDV逃逸宿主先天免疫中的作用。感染宿主细胞后,PEDV通过其NSPs和辅助蛋白涉及的机制拮抗宿主先天免疫反应,从而促进病毒逃逸宿主免疫防御。PEDV感染后,宿主细胞启动免疫信号级联,随后主要被病毒NSP家族蛋白和ORF3蛋白抑制。病毒识别通过细胞膜上的TLR发生,激活MyD88和TRIF等通路,而包括RIG-I和MDA5在内的细胞内PRR检测病毒RNA,触发MAVS介导的信号通路。PEDV的NSPs抑制IRF和NF-κB等关键信号分子,破坏IFN-I的产生和信号传导。此外,这些NSPs抑制核内抗病毒因子,包括ISG。此外,PEDV干扰MHC-I抗原呈递途径,进一步使NK细胞难以识别受感染细胞。总的来说,这些机制战略性地使PEDV能够逃逸宿主先天免疫系统。TLR:Toll样受体;TIRAP:Toll白介素1受体结构域衔接蛋白;TRAM:Toll样受体衔接分子;MyD88:髓样分化初级反应蛋白88;TRIF:含TIR结构域的衔接子诱导干扰素-β;IRAK4:白介素-1受体相关激酶4;TAB1:TGF-β激活激酶1和MAP3K7结合蛋白1;TAK1:转化生长因子-β激活激酶1;IKKβ:IκB激酶β;IKKα:IκB激酶α;IκBα:核因子κB抑制因子α;IKKε:核因子κB抑制因子ε激酶;ISG:干扰素刺激基因;STING:干扰素基因刺激物;ISRE:干扰素刺激反应元件。该图由BioGDP.com创建。
2.2.2 NSP2
FBXW7作为一种先天抗病毒因子,能增强RIG-I和TBK1的表达,同时诱导ISG以提高宿主的抗病毒状态。PEDV NSP2通过与FBXW7相互作用并将其靶向进行K48连接的泛素-蛋白酶体降解来抵消这种防御,从而抑制细胞抗病毒免疫(图2)。最近的研究进一步确定NSP2是一个靶向TBK1的毒力决定因子。从机制上讲,NSP2诱导巨自噬并招募选择性自噬受体NBR1(BRCA1基因邻接基因1),随后NBR1介导TBK1的K48连接泛素化并将其递送至自噬体进行降解。这确定了肠道冠状病毒NSP2是通过NBR1介导的TBK1选择性自噬来抑制先天免疫的主要调节因子。
2.2.3 NSP3
作为PEDV基因组编码的最大的跨膜蛋白,NSP3具有复杂的结构,具有两个木瓜蛋白酶样蛋白酶(PLpro)结构域,命名为PLP1和PLP2。先前的证据表明,PEDV在感染期间破坏IRF3激活,从而阻断双链RNA(dsRNA)诱导的IFN-β产生。在这种免疫逃逸范式中,PLP2通过其去泛素化酶(DUB)活性作为关键的IFN拮抗剂,该活性降低了受感染细胞中全局泛素化蛋白水平。此外,PEDV PLP2表现出严格的底物特异性——处理K48和K63连接的多聚泛素链,具有强大的DUB功能。
除了这些功能外,PEDV PLP2在先天免疫调节中表现出强大的抑制活性。它物理性地与RIG-I和STING相互作用(通过免疫共沉淀证明)并对这两个分子进行去泛素化。这种修饰强烈抑制了RIG-I和STING信号通路的激活,从而减少了IFN的产生。此外,PEDV编码的PLPro作为一种免疫逃逸效应因子,对抗ISG,从而削弱宿主抗病毒防御以促进持续性病毒复制(图2)。对SARS-CoV和TGEV NSP3的补充研究揭示了保守的机制,即NSP3通过抑制IκBα泛素化以及抑制p56磷酸化和核转位来阻断NF-κB介导的细胞因子反应。PEDV是否利用相同的途径进行免疫逃逸仍是一个有待进一步研究的开放性问题。
2.2.4 NSP5
NSP5在冠状病毒复制中作为一种必需的3C样蛋白酶(3CLpro),执行对病毒生命周期至关重要的蛋白水解加工。鉴于其在多聚蛋白成熟中不可替代的作用,该酶也被称为主要蛋白酶,其催化活性依赖于由Cys145和His41组成的催化二聚体。值得注意的是,NSP5在所有已知的冠状病毒中表现出异常的进化保守性,保持高度保守的氨基酸序列和三级结构。在生理条件下,该蛋白酶主要作为同源二聚体运作,精确切割病毒前体多聚蛋白pp1a/pp1ab。这种加工产生非结构蛋白NSP4-NSP16,它们共同支撑病毒结构完整性和复制功能。
NF-κB必需调节剂(NEMO)是宿主先天免疫中的关键分子,协调RNA病毒响应中NF-κB、IRF3和IRF7的激活,从而控制IFN的产生。先前的一项研究表明,NEMO不仅促进MAVS介导的IKKα/β激活,而且对于最佳的TBK1/IKKε磷酸化也是不可或缺的——这是先天免疫信号级联中的关键事件。
值得注意的是,PEDV通过其NSP5靶向NEMO来破坏宿主免疫防御。除了其作为负责病毒多聚蛋白内大部分切割的半胱氨酸蛋白酶的既定作用外,NSP5还作为一种有效的IFN拮抗剂,利用其蛋白水解活性在PEDV感染期间在谷氨酰胺231(Q231)处切割NEMO。这个切割位点在进化上是保守的,并严重破坏了IFN诱导所需的NEMO依赖性NF-κB激活。同时,NSP5靶向转录因子STAT2进行蛋白水解切割。这些双重切割事件破坏了NEMO介导的IFN诱导能力,并破坏了IFN-I信号级联,最终导致IFN-I产生显著减弱。这种多管齐下的逃逸策略促进了PEDV逃逸宿主免疫监视(图2)。
上述蛋白水解活性代表了PEDV采用的一种关键免疫逃逸策略。同时,PEDV NSP5通过抑制其TBK1上游的激活级联来破坏RIG-I/MDA5信号轴(图2)。关键的是,由于TBK1是连接病原体识别和IFN诱导的中心信号枢纽,这种上游阻断使NSP5能够在起始阶段破坏先天免疫反应。通过在最早的检查点中止抗病毒信号的启动,NSP5为病毒在宿主细胞内的复制、组装和传播创造了时间和空间优势。
2.2.5 NSP6
作为在PEDV感染期间协调宿主自噬的关键NSP,NSP6在劫持宿主细胞生理以促进病毒复制方面发挥着核心作用。在猪肠上皮细胞(IPEC-J2)中,NSP6通过下调mTOR及其下游效应因子p7056K的磷酸化来抑制PI3K/Akt/mTOR信号轴,从而诱导自噬体形成。关键的是,干扰内源性p53表达显著减弱了NSP6诱导的自噬并降低了病毒滴度,证实了NSP6介导的自噬依赖于PI3K/Akt/mTOR-p53轴,并且对于病毒在肠道细胞中的高效复制至关重要。
值得注意的是,NSP6的自噬诱导机制表现出细胞类型特异性。在Vero细胞中,PEDV感染不会引起mTOR磷酸化的显著变化;相反,NSP6通过激活AMPK-ULK1和JNK通路触发自噬,不依赖于PI3K/Akt级联反应。这种差异可能归因于信号通路基线活性的差异以及不同细胞类型中NSP6相互作用蛋白的不同表达谱,表明PEDV灵活地调整其自噬调节策略以优化在不同宿主微环境中的感染。
此外,作为冠状病毒复制-转录复合物(RTC)的一个组成部分,NSP6诱导的自噬不仅为病毒复制提供能量和底物,还降解宿主抗病毒蛋白(例如,IRF3,TBK1)以抑制先天免疫反应。这建立了一个协同的“自噬诱导-免疫抑制-病毒复制”循环,增强了PEDV逃逸宿主防御的能力。
2.2.6 NSP7
由83个氨基酸组成的NSP7在不同的PEDV毒株中表现出高度的序列保守性。亚细胞定位研究证实其在宿主细胞中主要具有细胞质功能。作为一种保守的病毒蛋白,NSP7在PEDV免疫逃逸中起着关键作用,特别是在破坏宿主IFN-I反应方面。通过量化启动子活性的双荧光素酶报告基因检测和基于qPCR的基因表达分析,我们证明NSP7剂量依赖性地抑制IFN-α诱导的IFN刺激反应元件(ISRE)启动子激活,从而抑制下游ISG表达(图2)。
NSP7通过多层机制拮抗IFN-I信号传导。它破坏IFN-α触发的JAK-STAT信号传导,减弱ISG表达。矛盾的是,免疫印迹分析表明,NSP7既不改变JAK1、Tyk2、STAT1或STAT2的蛋白丰度或磷酸化动力学,也不阻碍干扰素刺激基因因子3(ISGF3)复合物的组装。关键的是,NSP7通过竞争性结合STAT1并破坏其被核转运蛋白α1(KPNA1)——STAT蛋白主要的核输入衔接子——识别来阻断STAT1/STAT2的核转位(图2)。这种KPNA1的隔离取消了ISGF3的核运输,最终抑制了IFN-I信号传导和下游抗病毒基因表达,以促进病毒免疫逃逸。
此外,PEDV NSP7靶向MDA5并与其caspase募集结构域(CARD)相互作用,从而将MDA5与蛋白磷酸酶1(PP1)催化亚基PP1α和PP1γ隔离开。这种相互作用阻断抑制了MDA5 Ser828的去磷酸化,从而维持了MDA5的失活状态并损害了MDA5介导的IFN产生。
2.2.7 NSP10和NSP14
NSP14在冠状病毒家族中高度保守。所有同源物都具有双重催化活性:3'–5'外切核糖核酸酶(ExoN)校对和N7-甲基转移酶(N7-MTase)帽修饰。通过协同的ExoN介导的RNA错误校正和N7-MTase驱动的RNA帽甲基化,这种双功能酶确保了基因组复制的保真度——为稳定的病毒传播和准确的遗传信息传递提供了机制基础。
通过组合过表达和RNAi策略在人和猪细胞中对NSP14功能进行的系统评估一致显示,其剂量依赖性地降低了RIG-I蛋白水平而不改变mRNA丰度,表明存在翻译后调控。放线菌酮追踪实验表明,NSP14将RIG-I的半衰期从约6小时缩短至2小时。这种降解被蛋白酶体抑制剂MG132完全阻断,但不受溶酶体抑制剂影响,明确涉及泛素-蛋白酶体途径。免疫共沉淀、共定位和截短突变分析确定了NSP14的N端1–180氨基酸结构域与RIG-I的CARD之间存在直接结合,为降解提供了结构基础。在功能上,双荧光素酶报告基因检测显示,NSP4使RIG-I介导的IFN-β启动子活性降低了约70%。作为关键的PRR,RIG-I通常检测PEDV RNA以激活MAVS通路,从而促进IRF3磷酸化和核转位,诱导IFN-I产生并建立抗病毒状态。
然而,NSP14介导的RIG-I降解显著降低了宿主细胞识别PEDV RNA的能力,损害了MAVS信号激活,并破坏了IRF3依赖性IFN-I的转录起始。因此,宿主无法启动及时的早期抗病毒反应,这通过两种不同的机制起作用:(i)它减少了IFN-I诱导的ISG对病毒复制的直接抑制,这些ISG抑制病毒RNA合成并破坏病毒蛋白翻译;(ii)它减弱了IFN-I介导的旁观者细胞警报,阻止邻近细胞进入先发制人的抗病毒状态。总的来说,这些效应创造了一个关键的时间窗口和允许的环境,促进了PEDV在受感染组织内广泛的复制、组装和细胞间传播。这种级联最终破坏了宿主-病毒平衡,有利于病毒持续存在。并且,与野生型病毒相比,NSP14缺陷的PEDV突变体引发了2.3倍更高的IFN-β诱导,并表现出病毒滴度降低1个对数级。总的来说,NSP14通过靶向RIG-I进行蛋白酶体降解以抑制IFN-I通路来促进病毒免疫逃逸。
133个氨基酸的NSP10蛋白(≈14.6 kDa)采用精细解析的三级结构,其特征是N端反平行α螺旋对形成紧凑核心,中央不规则的β片赋予结构可塑性,以及C端结构域包含两个锌指基序和广泛的卷曲区域,共同支撑其功能。在冠状病毒RTC内,NSP10与NSP14、NSP16和其他复制酶亚基合作以维持基因组保真度。具体来说,NSP10在病毒RNA合成过程中变构刺激NSP14的3'-5'外切核糖核酸酶(ExoN)活性和NSP16的2'-O-甲基转移酶功能——增强复制准确性以确保精确的遗传信息传递。结构和生化分析证实,NSP10和NSP14持续相互作用形成异源二聚体复合物,结构域特异性结合稳定并增强了NSP14的催化效率。功能研究确定NSP10是病毒RNA合成的重要辅助因子,其与NSP14的结合将ExoN校对活性提高了>35倍,并加速了错误掺入核苷酸的切除。
2.2.8 NSP15
作为一种冠状病毒内切核糖核酸酶,NSP15在病毒RNA合成过程中利用其内切核酸酶活性靶向TBK1和IRF3 RNA进行降解。鉴于TBK1和IRF3在宿主IFN调节中的核心作用,其RNA水平的降低直接抑制了IFN产生,并显著减少了ISG诱导,从而使PEDV能够破坏宿主先天免疫反应(图2)。一项研究表明,残基H226A、H241A和K282A对于PEDV NSP15的核糖核酸酶活性至关重要。通过使用双荧光素酶报告系统进行大规模筛选,研究人员确定NSP15是poly(I:C)诱导的IFN-β和IFN-λ激活的有效抑制因子——这一发现pinpointed了支撑PEDV免疫逃逸的关键机制。随后使用具有严格对照的感染性克隆进行的反向遗传学研究表明,NSP15对宿主IFN-I(IFN-β)和IFN-III(IFN-λ)反应的拮抗作用严格依赖于其内切核糖核酸酶活性,明确确立了其功能的分子基础。
2021年,Gao等人证明传染性支气管炎病毒(IBV)NSP15通过减少宿主细胞中dsRNA的积累来逃避蛋白激酶识别并抑制SG形成。从机制上讲,EndoU核糖核酸酶活性被证明对SG抑制至关重要。对PEDV的平行研究表明,在LLC-PK1细胞中过表达NSP15显著抑制了eIF2α依赖性和非依赖性的SG组装。一致地,冠状病毒NSP15表达在多种细胞模型中普遍阻断了PKR-eIF2α-SG信号传导。鉴于催化核心结构域在冠状病毒中的高度保守性,这些发现表明NSP15介导了对宿主抗病毒反应的广泛保守的抑制。
2.2.9 NSP16
在冠状病毒复杂的基因表达和调控网络中,NSP16因其独特的功能性和特殊的保守性而成为病毒生命周期的重要组成部分。这种2'-O-甲基转移酶(MTase)在病毒RNA合成中起着关键作用。跨多个冠状病毒的序列分析显示,不同毒株间NSP16的氨基酸保守性异常高——表明其在病毒生存和传播中具有不可或缺且进化上保守的功能。
关于PEDV介导的IFN拮抗作用的研究表明,NSP16在病毒-宿主相互作用中作为一种有效的免疫逃逸因子。从机制上讲,NSP16利用其甲基转移酶活性抑制IRF3磷酸化——这是先天免疫信号传导中的一个关键节点。受损的IRF3磷酸化直接阻断了RIG-I/MDA5触发的信号级联(图2),使PEDV能够逃逸宿主免疫监视并对抗抗病毒反应。结构和功能分析进一步证明,NSP16保守的KDKE基序是抑制IFN-β和ISRE启动子激活的关键结构域,为其免疫颠覆功能提供了机制性见解。将催化天冬氨酸残基突变为丙氨酸会消除甲基转移酶活性,显著削弱这些免疫颠覆功能。
研究表明,NSP16显著下调RIG-I/MDA5介导的IFN-β和ISRE活性,从而减弱宿主抗病毒免疫。相应地,NSP16过表达显著抑制了IFIT家族成员(IFIT1,IFIT2,IFIT3)的mRNA水平——这些是关键的抗病毒效应因子,其受抑制的表达证实了NSP16的免疫抑制功能。值得注意的是,在PEDV的调控网络中,NSP10增强了NSP16介导的IFN-β抑制,揭示了协调的免疫逃逸。
作为IFN-I产生途径中的核心转录因子,IRF3需要磷酸化才能进行核转位并与IFN-β启动子结合。NSP16直接抑制IRF3磷酸化,从而阻止其入核和随后的IFN-β基因转录,这显著减少了宿主细胞分泌IFN-β。从病毒的角度来看,降低的IFN-β水平减轻了复制过程中的免疫压力:病毒RNA聚合酶逃脱了IFN-β诱导的ISG的抑制,使得病毒基因组能够高效复制,而病毒蛋白翻译则不受ISG干扰,提高了病毒粒子组装效率。相反,从宿主的角度来看,不足的IFN-β无法激活邻近细胞中的抗病毒基因表达,使它们容易受到PEDV感染,并扩大了病毒在宿主体内的传播。此外,NSP16介导的RIG-I/MDA5驱动的IFN-β激活下调破坏了宿主涉及多个PRR的双重防御机制,促进了病毒在宿主细胞中的稳定定植,并促进了随后的病毒粒子释放和二次感染。这种协调的抑制例证了一种进化出的病毒策略,用于破坏先天免疫,其中NSP16在为PEDV持续存在创造允许的细胞内环境中发挥了关键作用。两种甲基转移酶NSP14和NSP16都拮抗先天免疫;然而,比较分析表明NSP16更有效地调节免疫相关基因表达并精确调控宿主反应。
2.2.10其他NSPs
PEDV NSP6通过PI3K/Akt/mTOR信号通路诱导自噬并促进病毒在猪IEC中的复制。作为冠状病毒RNA复制机制的关键组成部分,NSP8与NSP7形成十六聚体复合物,介导核酸与NSP12的结合,构成最小的RNA聚合酶复合物。NSP8仅在相同温度下与NSP7和NSP12组装时才表现出高的RNA依赖性聚合酶活性。此外,PEDV NSP8在体外通过降低IRF1启动子活性来抑制IFN-III活性。使用感染性克隆的重构研究尝试——但未能——在重排NSP7和NSP8基因后拯救出有活力的病毒。这项工作以及积累的证据确立了NSP7和NSP8作为病毒复制必需的辅助因子,表明它们在PEDV介导的先天免疫抑制中的潜在作用,尽管其机制细节尚未解决。
PEDV的非结构蛋白作为先天免疫逃逸的核心效应分子发挥作用,每种蛋白靶向宿主抗病毒信号级联的不同节点。总的来说,它们构成了一个协调的网络,不仅抑制IFN产生和阻断IFN信号传导,还逃避PRR的检测。这种多方面的策略对于PEDV在宿主细胞中建立持续性感染至关重要。
2.3辅助蛋白ORF3在PEDV逃逸宿主先天免疫中的作用
作为PEDV中唯一已鉴定的辅助蛋白,约25 kDa的ORF3关键地调节病毒毒力和复制。ORF3主要定位于细胞质,部分分布于ER和高尔基体。它在受感染细胞的核周区域和囊泡结构中与S蛋白共定位并相互作用,共同调节病毒复制。ORF3抑制IFN-I(IFN-β)和IFN-III(IFN-λ1)的产生,尽管其精确的抑制机制有待进一步验证。Ye等人建立了稳定表达ORF3的Vero细胞来研究其亚细胞定位和宿主相互作用,同时评估其对病毒粒子产生的影响。结果显示ORF3定位于细胞质,延长S期从而破坏细胞周期进程,并且与天然Vero细胞相比增加了囊泡形成。值得注意的是,减毒PEDV在表达ORF3的细胞中比强毒株复制更有效。
此外,ORF3通过细胞凋亡和自噬途径对病毒复制施加双重调节:它直接抑制受感染细胞的凋亡以增强病毒增殖,同时促进LC3-I向LC3-II的转换以诱导自噬流。ORF3在ER中积累,上调GRP78表达并激活PERK-eIF2α通路,从而触发ERS。这启动了未折叠蛋白反应(UPR)——一种恢复蛋白质稳态的保守机制——最终诱导细胞死亡和自噬。ORF3还通过调节NF-κB信号传导来拮抗宿主先天免疫,具体通过:(i)抑制IκBα和核因子p65的磷酸化,以及(ii)损害p65核转位(图2),这共同减少了促炎细胞因子的产生(例如,IL-6,IL-8)。矛盾的是,虽然ORF3增强了IκBβ介导的NF-κB启动子活性,但它反直觉地抑制了IκBβ驱动的IFN-β启动子激活和mRNA表达。过表达实验证实了ORF3抑制poly(I:C)诱导的IFN-I产生的能力。
3.自噬对PEDV逃逸宿主先天免疫的影响
病毒对靶细胞的感染通常诱导细胞自噬和程序性细胞死亡。虽然病毒利用宿主ER折叠新生的病毒蛋白,但未折叠蛋白的过度积累会触发ERS,从而诱导自噬。Lin等人证明,用100 nM雷帕霉素(一种诱导自噬的mTOR通路抑制剂)处理PEDV感染的IPEC-J2细胞,可增强PEDV复制,证实了自噬的促病毒作用。
3.1 PEDV通过ERS诱导自噬
ERS是一种关键的细胞应激反应机制,其中UPR处理ER腔内的错误折叠蛋白。这个质量控制系统在检测到蛋白质错误折叠时启动多方面的调节措施,包括减弱进一步蛋白质向ER的转运以防止腔内过度拥挤——从而维持蛋白质稳态。从病毒学角度来看,病毒通常利用宿主ER在合成后折叠新生的病毒蛋白。然而,ER腔内未折叠蛋白的过度积累通过超载诱导的信号传导触发ERS。细胞自噬——一种溶酶体依赖性降解途径——构成了另一个可由多种刺激(包括ERS)诱导的基本生物过程。在ERS激活期间,自噬被上调以消除异常蛋白质和受损细胞器,从而维持细胞稳态。
在一系列关于PEDV感染机制的研究中,Xu等人首先用PEDV感染宿主细胞,随后检测到病毒结构蛋白E和N在ER内大量积累。这一观察促使研究这些基本结构成分的亚细胞定位是否与宿主生理变化相关。为了解决这个问题,该团队设计了检查E/N蛋白与ERS之间关系的实验。结果表明,这两种蛋白都显著诱导宿主细胞中的ERS——这一结论得到了多个实验数据集的强有力支持。鉴于ER在蛋白质合成、加工和功能稳态中的关键作用,这些发现表明在PEDV感染期间,E和N蛋白可能通过触发ERS破坏ER功能,从而损害细胞生理学。这一机制为PEDV如何操纵宿主细胞过程以促进感染提供了关键见解。
Zou等人揭示了一个值得注意的机制,即在PEDV感染期间主要定位于ER的ORF3蛋白通过升高GRP78表达来激活PERK-eIF2α信号轴,从而诱导ERS。此外,这种病毒蛋白通过促进LC3-I向LC3-II的转换(这是自噬体形成的关键步骤)来触发自噬。关键的是,使用4-PBA药理学抑制ERS显著抑制了LC3脂化,证明ORF3诱导的自噬在机制上依赖于ERS激活。
3.2 PEDV相关蛋白对自噬的调控
除了NSP6介导的通过自噬依赖性逃逸先天免疫的宿主免疫通路调节外,PEDV还部署多种病毒蛋白——包括N蛋白、NSP2和ORF3——通过不同的机制利用自噬。N蛋白诱导ERS,间接触发自噬,同时利用此过程降解抑制IFN-β产生的抗病毒宿主蛋白(例如,IRF3,TBK1)。NSP2招募选择性自噬受体NBR1来介导TBK1的自噬降解,从而瓦解关键的先天免疫信号枢纽。ORF3激活PERK-eIF2α轴以驱动ERS相关的自噬,该过程严格依赖于GRP78上调。总的来说,这些蛋白通过靶向自噬通路的不同节点,协调了一个协同的自噬依赖性免疫逃逸网络。然而,它们之间的精确相互作用和信号串扰需要进一步研究,以阐明PEDV对宿主防御的系统性破坏。
4.PEDV通过隐藏PAMP逃逸宿主先天免疫
在病毒-宿主相互作用过程中,病毒进化出复杂的逃逸策略以保持基因组完整性并确保生产性感染。关键策略包括病毒内切核糖核酸酶活性和5'帽结构,这些可以保护病毒RNA免受宿主识别和降解。此外,非结构蛋白NSP14的N7-MTase活性对于病毒转录/翻译是不可或缺的,同时它通过将病毒mRNA伪装成“自身”来破坏宿主防御——特别是阻止PRR启动针对病毒mRNA的先天免疫反应。
PEDV NSP15具有内切核糖核酸酶活性,使其能够通过切割病毒dsRNA来实现免疫逃逸,从而减少积累并防止被宿主传感器MDA5和PKR识别。关键的是,NSP15的EndoU催化功能表现出情境依赖的可塑性——受dsRNA二级结构和转录后修饰的调节——能够动态适应细胞内条件。这种结构灵活性允许NSP15抑制dsRNA传感器的激活,最终促进持续性病毒复制。PEDV NSP16,一种甲基转移酶家族酶,催化病毒RNA帽修饰。
这种生化模拟使病毒RNA在结构上与宿主mRNA无法区分,有效地逃避了MDA5的监视并减少了抗病毒免疫激活。在RNA病毒复制过程中,不可避免地产生dsRNA和5'-三磷酸RNA,通常会触发PRR介导的免疫反应。然而,通过包括NSP15和NSP16在内的NSPs的协同活动,PEDV破坏了宿主免疫监视,确保持续的病毒复制和传播。
5.PEDV逃逸宿主先天免疫的其他途径
MAPK信号通路通过关键介质ERK、JNK和p38协调细胞对细胞内和细胞外刺激的反应。在PEDV感染期间,ERK和p38的激活反常地增强了病毒复制,而不诱导宿主细胞周期阻滞或细胞凋亡。Guo等人证明,PEDV利用泛素-蛋白酶体途径降解磷酸化的STAT1(p-STAT1),从而抑制IFN信号传导。此外,感染触发caspase-8介导的Ras-GTP酶激活蛋白结合蛋白1的切割,并破坏SG组装以促进病毒复制。PEDV还通过蛋白酶体依赖性机制降解分区缺陷蛋白3(PARD3)——一种维持上皮紧密连接的支架蛋白——以促进病毒增殖。关键的是,HSP27通常通过激活NF-κB来诱导IFN-β和下游ISG,从而增强抗病毒反应。然而,PEDV在Marc-145细胞中抑制HSP27功能,减弱IFN-β和ISG表达以逃逸宿主免疫。
6.结束语
近年来,关于PEDV逃逸宿主先天免疫系统已取得显著进展。研究揭示了PEDV采用多种复杂机制来破坏宿主免疫监视或拮抗先天免疫反应。这些包括直接或间接抑制宿主IFN产生、隐藏其PAMP以避免被宿主PRR识别,以及通过NF-κB信号传导等途径减弱炎症反应。此外,PEDV利用宿主细胞生理过程,包括自噬、ERS、细胞凋亡和各种宿主细胞信号通路,以促进病毒复制和逃逸先天免疫。
虽然当前研究揭示了PEDV免疫调控的部分机制,但该领域仍存在显著的知识空白,这同时描绘了未来研究的有希望的方向。一方面,不同免疫逃逸机制在体内的层级贡献尚未确定。例如,在自然感染期间,E蛋白介导的ERS、IL-8表达调控、NF-κB信号调节或其他潜在未知策略是否在病毒感染和传播中起主导作用,需要通过动物模型中的动态追踪实验来验证。另一方面,病毒蛋白之间是否存在协同或拮抗效应(例如,NSP10增强NSP14活性以协同抑制IFN反应),从而形成更高效的免疫逃逸网络,这是一个未解的难题。此外,宿主异质性对免疫逃逸的影响仍不清楚——PEDV的逃逸效率是否会因宿主年龄、品种或不同感染部位导致的免疫分子(如RIG-I和IRF3的表达水平)差异表达而改变?这些问题的答案需要在未来研究中建立更真实的感染模型。
从技术角度来看,多组学整合分析可能是一个关键的解决方案。通过结合转录组学、蛋白质组学和代谢组学数据,这种方法可以系统地识别PEDV感染后宿主免疫通路中的核心调控节点,同时发现新的病毒-宿主相互作用靶点。超分辨率成像技术(例如,STED,SIM)将能够直接可视化病毒蛋白与宿主分子之间的原位结合动力学,阐明亚细胞定位对免疫调节的机制性影响。此外,基于已知相互作用位点的靶向干预实验可以功能性验证关键机制,为开发新的预防和控制策略提供直接证据。
总之,未来的研究应采用“机制阐明—技术验证—转化应用”的框架。通过不断发现和解决未解问题,不仅可以加深我们对PEDV如何突破宿主先天免疫屏障的理解,还可以为设计更有针对性和适应性的对策(如多表位疫苗和免疫调节剂)奠定基础。最终,这些努力将有助于保障动物健康,促进畜牧业的可持续发展。
猪流行性腹泻病毒如何逃逸宿主先天免疫?
来源:华派
发布时间:2025-11-26
浏览量:2396




